Defects in the hepatocyte nuclear factor 1-β (HNF1β) gene result in the development of a constellation of defects including cystic renal disease, maturity-onset diabetes of the young (MODY), and genitourinary abnormalities; and the less common findings include agenesis of the pancreas and liver abnormalities. We report a young girl with an HNF1β mutation (variant c.479T > G (p.M160R)), who had a history of neonatal cholestasis, persistent liver dysfunction, and developed insulin-dependent diabetes without renal involvement. Further, we review the literature related to the hepatic involvement associated with HNF1β mutations.
Hepatocyte nuclear factor-1β (HNF1β) is a member of the homeodomain-containing family of transcription factors and is encoded by the HNF1β gene located on chromosome 17 [1,2]. HNF1β is involved in the transcriptional and functional regulations of the liver, biliary system, kidney, urogenital tract, and pancreatic β-cells [3,4]. Defects of this gene have been described in a small subgroup of patients with MODY, including point mutations and whole gene deletions, with the majority being de novo whole gene mutations [5,6]. The phenotypical spectrum of individuals with HNF1β mutations varies widely. The major features are MODY and cystic renal disease, however, agenesis of the pancreas, urogenital malformations, hyperparathyroidism, gout, cognitive delay, autism spectrum disorder, and hepatic disorders have also been described [3,4].
An African-American female presented at 6-weeks of age with poor growth and jaundice, which, as the mother stated, had been present since birth. She was born to a prima gravid mother at term via spontaneous vaginal delivery, following an uncomplicated pregnancy. At presentation, the infant was having pigmented bowel movements. The only abnormal physical findings were jaundice, scleral icterus, a grade 2/6 soft systolic murmur consistent with a Still’s murmur, and mild hepatosplenomegaly. Laboratory examination showed evidence of cholestasis with elevated levels of transaminases (alanine-amino transaminase, ALT = 110 U/L), total and direct bilirubin (11.8 mg/dl and 8.5 mg/dl, respectively), gamma glutamyl transferase (GGT, 150 U/L), and serum bile acids. Serum albumin and prothrombin time were normal. Further testing for an etiology was non-diagnostic, including serum amino acids, serologic testing for human immunodeficiency virus, toxoplasmosis, cytomegalovirus, rubella, syphilis, and alpha-1-antitrypsin phenotype. Ultrasound of the liver and gallbladder was normal. Liver biopsy showed mild bile ductule proliferation in portal areas with mild inflammatory cell infiltrate, no fibrosis, and rare foci of extramedullary hematopoiesis.
The patient maintained normal hepatic function, as evidenced by normal serum albumin and prothrombin time. Bilirubin normalized at 5 months of age, however, serum levels of transaminases and GGT remained elevated, fluctuating over time (Figure 1). Repeat liver biopsy at 15-months of age showed patchy minimal chronic lymphocytic inflammation of the portal areas and patchy fibrosis with portal-to-portal bridging. A third liver biopsy was performed at 6.25 years of age to assess for worsening fibrosis due to persistence of biochemical evidence for hepatobiliary injury and the presence of fibrosis on the previous biopsy. The biopsy showed unremarkable portal tracts with no inflammation, fibrosis, or bile ductular proliferation.
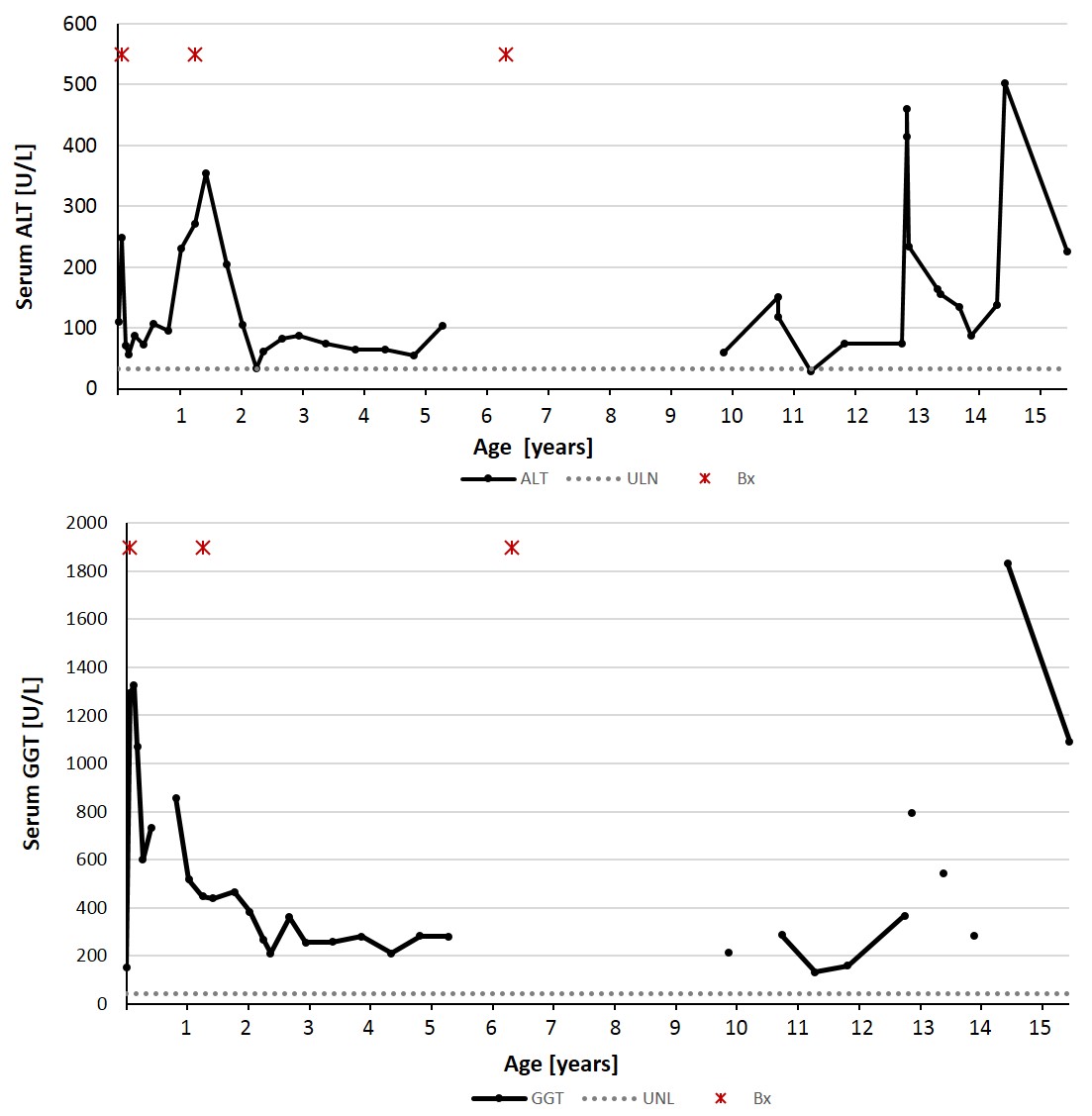
Figure 1. The levels of serum ALT and GGT fluctuated during a period of 15 years. Abbreviations: ALT, alanine-amino transaminase; GGT, gamma glutamyl transferase.
At 11-years of age, she presented with polyuria, polydipsia, and polyphagia. Laboratory evaluation showed ketonuria, metabolic acidosis, and hyperglycemia, consistent with a diagnosis of diabetic ketoacidosis. Antibodies to glutamic acid decarboxylase and islet cells were negative. A diagnosis of diabetes mellitus was rendered and treatment initiated with insulin replacement. At this point, it was elected to look for mutations in the HNF1β gene to explain the association between the persistent hepatic disease and the early onset insulin-dependent diabetes mellitus. HNF1β gene sequencing detected two gene defects: c.721G > A; p.A241T and c.479T > G; p.M160R. Due to the known association of renal disease with HNF1β gene defects, the patient underwent testing of renal function and a renal ultrasound, which were normal.
At her most recent follow-up at 17-years of age, she continues to have fluctuating levels of transaminases (ALT = 181 U/L; AST = 163 U/L) and GGT (1147 U/L); her diabetes is under poor control as evidenced by an elevated hemoglobin A1c (14.0%; upper limits of normal is 6.1%). She is presently receiving ursodiol (300 mg BID) and rifampin (300 mg BID) for control or pruritus and remains asymptomatic regarding liver disease.
More than 100 different mutations in the HNF1β gene have been reported to cause MODY [7]. The difference in clinical presentation of the HNF1β mutation is thought to be caused by the differences in HNF1β activity in conjunction with the repression of HNF1α activity in selected promoters and tissues [8]. Raile et al, suggested that gene dosage of other genetic factors and/or environmental triggers could play a role in the pattern and severity of the HNF1β-associated MODY [9]. Due to the rarity of patients with HNF1β mutation, the clinical manifestations and correlation with gene mutations are not well understood.
To date, 47 individuals have been reported with liver disease associated with HNF1β gene defects, 25 as case reports [6,8-14] and 22 identified in patients with the known renal disease being screened for HNF1β defects [15-17]. Liver involvement may present as cholestasis, elevated liver enzyme levels (ALT and/or GGT), hepatomegaly, or steatosis.
Clinical cholestasis manifested as jaundice has been reported in both children and adults with HNF1β defects. Overall, eight of 25 reported patients (32%) presented with cholestasis. Five children, including our patient, have been reported, all identified during the neonatal period. An etiology for cholestasis was not established in four, while one child was diagnosed with biliary atresia and underwent portoenterostomy [8- 10,13]. Long-term follow-up of three patients showed resolution of jaundice between 5 and 12 months of age. The diagnosis of an HNF1β defect was not entertained until the children developed insulin-dependent diabetes mellitus [8-10,13]. The exception is the patient described by Kotalova et al, where a defect was found in the HNF1β gene as part of a study looking for genes associated with biliary atresia [13]. In a case series of three adults, cholestasis was seen in association with poorly controlled diabetes and secondary renal insufficiency [14]. Electron microscopy showed paucity or absence of cholangiocyte cilia in these patients, suggesting a causative role in cholestasis [14].
Persistent and fluctuating liver enzymes are common findings in patients with defects in HNF1β. The percentage of patients from case reports with biochemical cholestasis (elevated GGT) is 78%, while the percentage with elevated ALT is 76% [6,8-14]. This is probably an over-representation as these patients had known liver disease and were subsequently found to harbor a defect in HNF1β. When searching for hepatic injury in patients with known HNF1β defects, the percentages are lower, ranging from 17% to 56% for GGT and 12% to 56% for ALT [14-18]. The degree of enzyme elevation varies from mild (twice the upper limit of normal) to severe (10-times the upper limit of normal).
Hepatomegaly has been found in 25% of the patients with an HNF1β defect when the liver size was commented on in the physical examination [6,8-10,12]. Steatosis has been reported in four patients, diagnosed by imaging studies and/or liver biopsy.
Liver biopsies performed during the evaluation of cholestasis or persistent elevation in liver enzymes in patients with HNF1β defects showed varied histological findings. In the cholestatic neonate with an HNF1β defect, liver biopsies have shown paucity of bile ducts, bile ductule proliferation, extramedullary hematopoiesis, severe biliary stasis, and mild to severe periportal fibrosis [8-10,13]. Liver biopsies in adults with HNF1β defects and cholestasis have shown non-specific findings of steatosis and sinusoidal dilatation [14]. Liver biopsy performed for persistent elevation in liver enzymes in an infant showed mild steatosis and extramedullary hematopoiesis [12], while the liver biopsies performed in adults showed mild cholestasis and normal histology [11]. It appears that the histologic findings are more pronounced in infants when compared to adults, suggesting that there may be an improvement in liver injury over time. Support for this concept is found in a previous report, as well as in our patient. Improvement in liver histology was reported in an infant by repeat biopsy at 21-months of age [10], while our patient had normalization of liver histology at 6.25-years of age.
HNF1β mutations affect multiple organs and the resulting defects may be subtle or subclinical, making the diagnosis based on clinical features difficult. There should be a high index of suspicion for an HNF1β gene mutation in children with early onset diabetes and a past history of neonatal cholestasis or persistent elevation in hepatic enzymes.
Received date: September 30, 2017
Accepted date: November 13, 2017
Published date: March 19, 2018
© 2018 The Author(s). This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC-BY).
Ref no 17 is not as per format.
ResponseThis has been corrected.
The authors presented a patient with HNF1b mutation who developed neonatal cholestasis, persistent liver dysfunction and IDDM and review of hepatic involvement of cases of HNF1b mutations. The paper is well-written and has interesting findings.
Accepted for publication
The manuscript has been revised as per changes recommended. It is accepted for publication.
The paper is well-revised and has interesting findings, I accept for publication
Camacho SM, Nowicki MJ. Hepatocyte nuclear factor 1-β gene mutation: Brief report and review of hepatic involvement. Ann Case Rep Images 2018;1(1):2. https://doi.org/10.24983/scitemed.acri.2018.00054
 PDF
PDF