Objective: To present a case and systematically evaluate trends in clinical presentation, imaging, histopathology, and management modalities and outcomes of all head and neck juxtacortical chondrosarcoma (HNJCS) cases reported in the existing literature.
Methods: We describe a rare case of HNJCS from our tertiary referral center following which the PubMed, MEDLINE, Embase, and Web of Science databases were searched for all HNJCS reports. Relevant titles and abstracts were screened. Resulting full-text articles were assessed for eligibility, and remaining studies were included for data extraction, summarization, and analysis.
Results: Potential studies were identified dating from May 1946 to February 2019. A total of nine cases were included in our systematic review, eight of which were identified from full-text articles and one recruited from our tertiary referral center. The median presentation age was 41 years with a 66.7% male preponderance. The commonest presenting sign was a painless, isolated swelling after a median symptom duration of 2.5 months. CT imaging revealed hypodense lesions with peripheral enhancement and micro-calcifications. T1-weighted MRI showed hypo- to iso-intense, lobulated masses with peripheral and/or septal enhancement. The masses were diffusely hyper-intense on T2-weighted MRI. Histopathology demonstrated septated lobules of malignant hyaline cartilage with a peripheral fibrous capsule. Most tumors were low- or intermediate-grade tumors with average diameter of 4.3 cm. Local recurrence was identified in only one case (four years after initial resection). No distal and/or nodal metastases were identified. All tumors were managed by wide- or narrow-margin surgical excision. Two reports employed adjuvant treatment. There was no evidence of disease at final follow-up (median of 1.5 years).
Conclusion: To the best of our knowledge, only nine cases of HNJCS have been adequately described. HNJCS have relatively consistent clinical and diagnostic profile regardless of location in the body. Surgical management yields excellent outcomes with low recurrence rates.
Chondrosarcomas (CS) comprise a diverse category of locally-aggressive cartilaginous malignancies and are the third most frequent primary malignancy of bone behind multiple myeloma and osteosarcoma [1]. CS have largely been documented in the long bones of the appendicular skeleton (femur and humerus), pelvis, and thoracic bones [2,3]. In the rare event that these tumors are found occupying the head and neck region, the most commonly affected subsites include the skull, facial bones, and laryngeal cartilage [3,4].
The chondrosarcoma family of malignancies encompasses several sub-types. Conventional chondrosarcoma (CCS), which can be primary or secondary (from benign cartilage tumors such as chondroma or osteochondroma), is seen in 85-90% of documented cases [1,5]. They can further be distinguished as central (if derived from the medullary cavity) or peripheral (if derived from the outer cortical bone) [1]. The other variant subtypes are uncommon and include mesenchymal, clear cell, myxoid, dedifferentiated, and juxtacortical (parosteal, periosteal) [6]. Of these, juxtacortical chondrosarcomas (JCS) have the rarest occurrence, accounting for 2% or less of all CS cases [2,6]. JCS are defined as malignant hyaline cartilage tumors, which occur on the surface of bone [7]. They are typically low or intermediate grade malignancies that originate from the periosteum, firmly distinguishing them from primary CCS [7]. JCS are usually found on the metaphysis or diaphysis of the femur and humerus [8-10], and occurrence in the head and neck region is exceedingly rare. These tumors appear to have a positive prognosis with low rates of recurrence, metastases, and a 5-year survival >90% for those with adequate surgical margins [8,10]. Single-modality therapy utilizing wide en bloc resection remains the treatment modality of choice, with adjuvant radiotherapy or chemotherapy having a limited role in management [8-10].
Our study reports a case of HNJCS in a 41-year-old male after which we systematically review, summarize, and analyze the findings from all HNJCS cases reported in literature.
Case Report
A 41-year-old male presented to our tertiary referral center for an incidental mass found on imaging after sustaining facial trauma. A head and neck computed tomography (CT) with contrast revealed a 2.5 x 3.0 cm heterogeneous, hypodense lesion in the left infratemporal fossa region at the mandibular notch (Figures 1A,B). Minor punctate calcifications were noted along the medial margin, but no osseous remodeling or periosteal reaction was detected. On retrospective comparison, the mass was also noted on a head and neck CT scan from 2014 as an undiagnosed mass measuring 1.8 x 1.6 cm. On office interview, the patient complained of constant, dull, left-sided midfacial pain with intermittent spikes of intensity over the past seven years, previously diagnosed as trigeminal neuralgia by his primary care physician. He denied any masticatory pain, trismus, dysphagia, odynophagia, dysphonia, dyspnea, hemoptysis, otalgia, or neck mass. His past medical history was notable for hyperlipidemia, type 2 diabetes mellitus, hypertension, morbid obesity, and resolved stage IA Hodgkin’s lymphoma of the left neck managed with chemotherapy four years prior. He had a 13-pack-year smoking history with occasional alcohol use. He denied any family history of cancer or head and neck tumors. Physical examination was unremarkable without evidence of a palpable mass or lymphadenopathy. There was full range of motion at the temporomandibular joints.
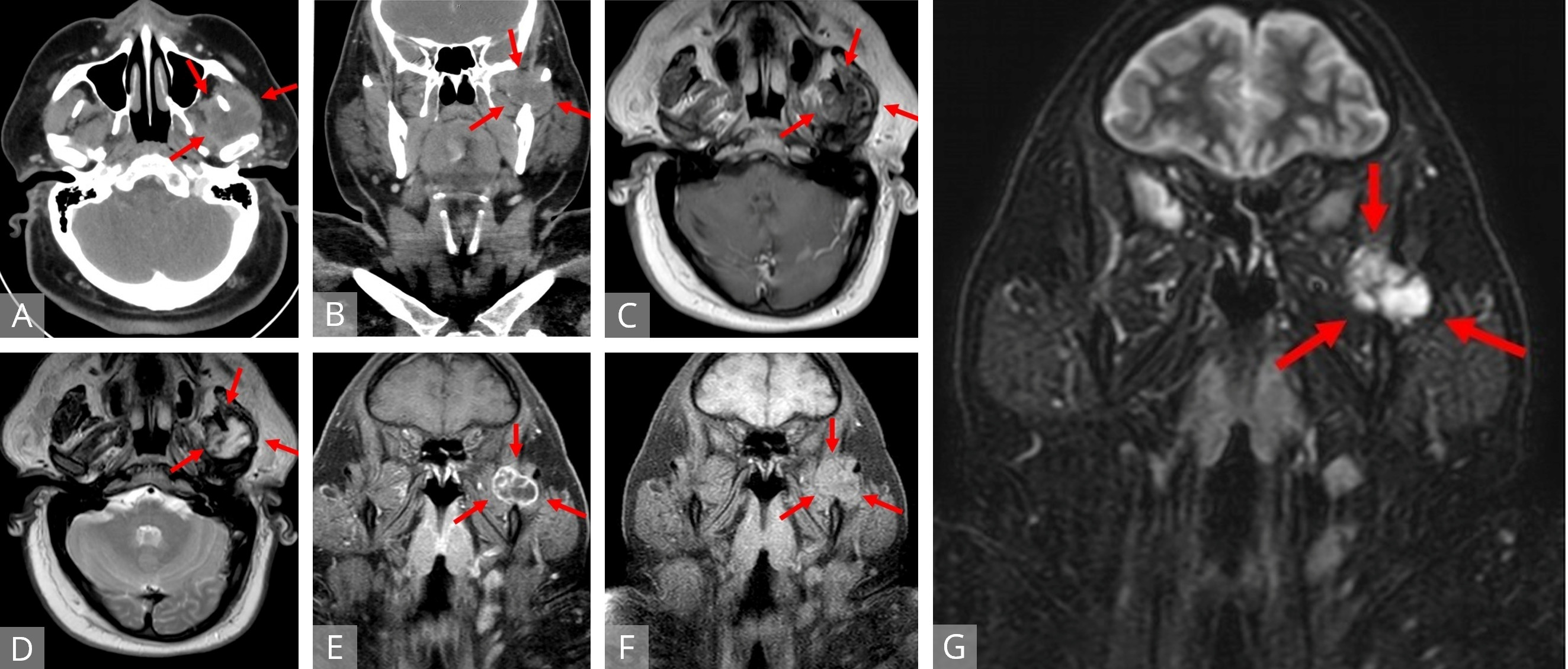
Magnetic resonance imaging (MRI) of the head and neck with intravenous contrast (Figures 1C-G) revealed a well-defined mass in the sigmoid notch of the left mandible appearing isointense on T1-weighted (T1W) imaging and heterogeneously hyperintense on T2-weighted (T2W) imaging. On fat-suppressed, post-contrast T1W imaging, the lesion exhibited peripheral and septonodular enhancement with clear lobulations. Cervical lymphadenopathy or osseo-muscular invasion were not identified.
A repeat CT-guided fine needle biopsy at our institution revealed a cartilaginous neoplasm with focal areas of increased cellularity and nuclear hyperchromatism. The working differential diagnosis prior to surgery was chondroma versus low-grade chondrosarcoma.
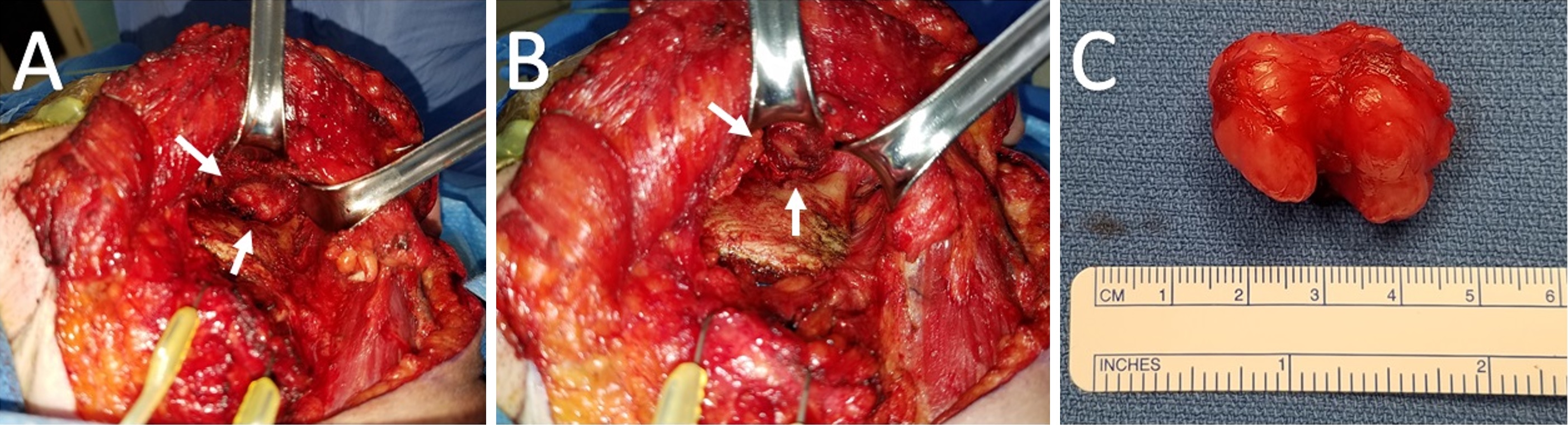
Subsequent tumor resection was performed via transcervical approach and coronoidectomy. The tumor was seen centered in the infratemporal fossa above the sigmoid notch, cradling the posterior portion of the coronoid process (Figure 2A). Extracapsular dissection was performed with complete resection of the tumor (Figure 2B).
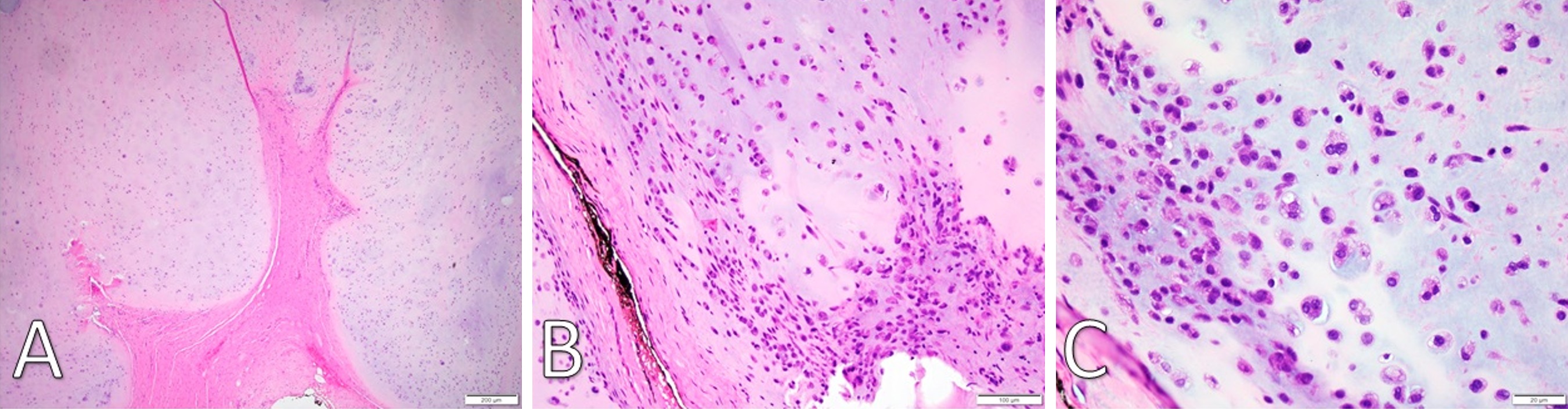
Gross examination revealed a 3.4 x 2.9 x 2.7 cm, exophytic, irregular, firm-elastic mass with a smooth lobulated, tan to red-brown external surface enveloped in a fibrous, adherent capsule (Figure 2C). On H&E histological examination at low power (4x), it was well-demarcated with a fibrous capsule and septations, forming lobulated areas of hyaline cartilage (Figure 3A). The fibrous capsule appeared to be continuous with the underlying periosteum. Under high power light microscopy (20x and 40x), areas of increased cellularity were identified within the hyaline cartilage. The fibrous septations lacked vascular infiltration and were partially invaded by spindle-shaped chondroid cells. The tumor cells were often found within lacunae and had marked hyperchromatism and nuclear pleomorphism with some binucleate forms. Spindle cells were also identified within the hyaline cartilage (Figures 3B,C). The final diagnosis was low-grade, T1N0Mx juxtacortical chondrosarcoma. All surgical margins were negative for malignancy. The patient’s postoperative course was unremarkable, and he was discharged home on the first postoperative day. No further adjuvant treatment was recommended. The patient was alive and without evidence of disease nine months postoperatively.
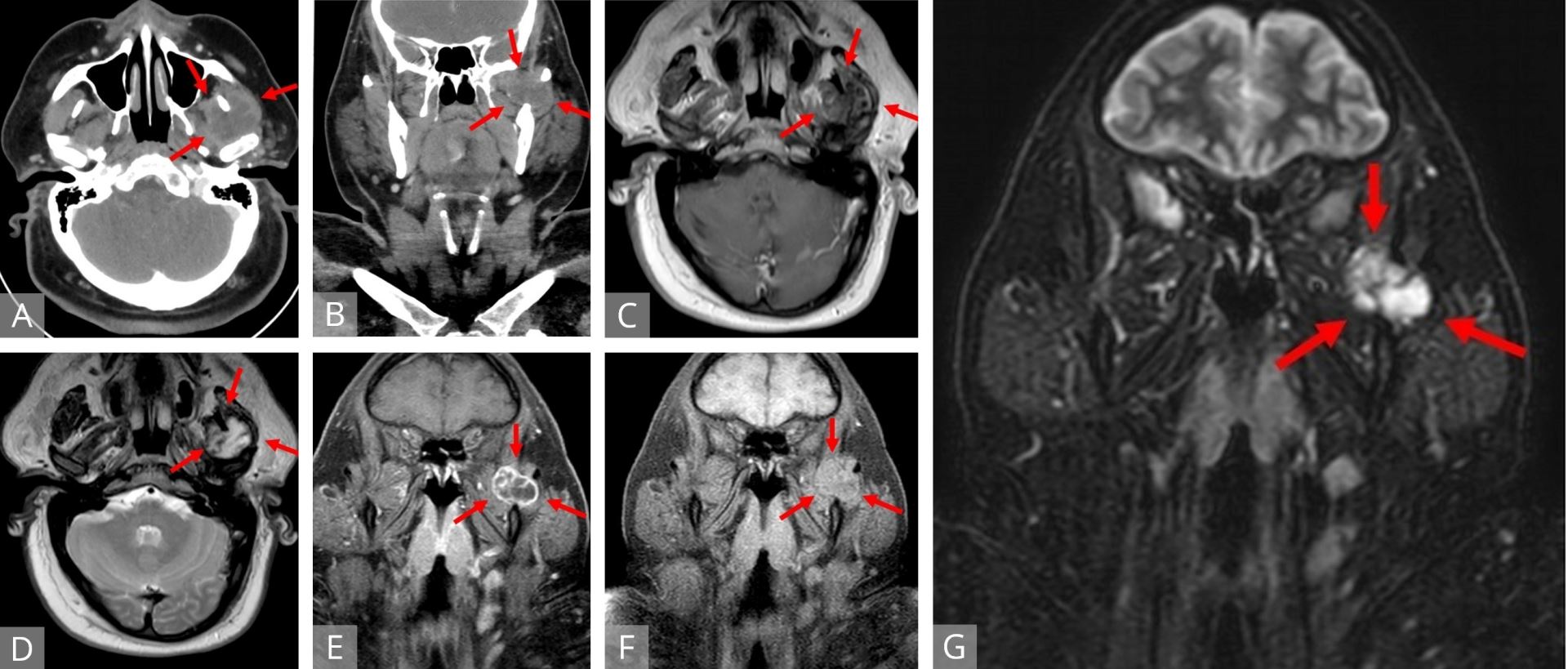
Figure 1. Axial (A) and coronal (B) CT of a heterogeneous, hypodense lesion in the left infratemporal fossa. Axial T1W (C) and T2W (D) MRI displaying a mass located between the left coronoid process and mandibular condyle with portions encircling the coronoid medially and laterally. The mass demonstrates peripheral isointensity with central heterogeneous hypointensity on T1 and central, streaking and variable hyperintensity on T2. Fat-suppressed coronal T1W MRI with (E) and without (F) contrast revealed an isointense, lobulated mass to surrounding musculature with peripheral and septonodular enhancement. Coronal STIR MRI (G) identifying a distinct lobulated tumor. MRI, magnetic resonance imaging; STIR, short tau inversion recovery; T1W, T1 weighted; T2W, T1 weighted.
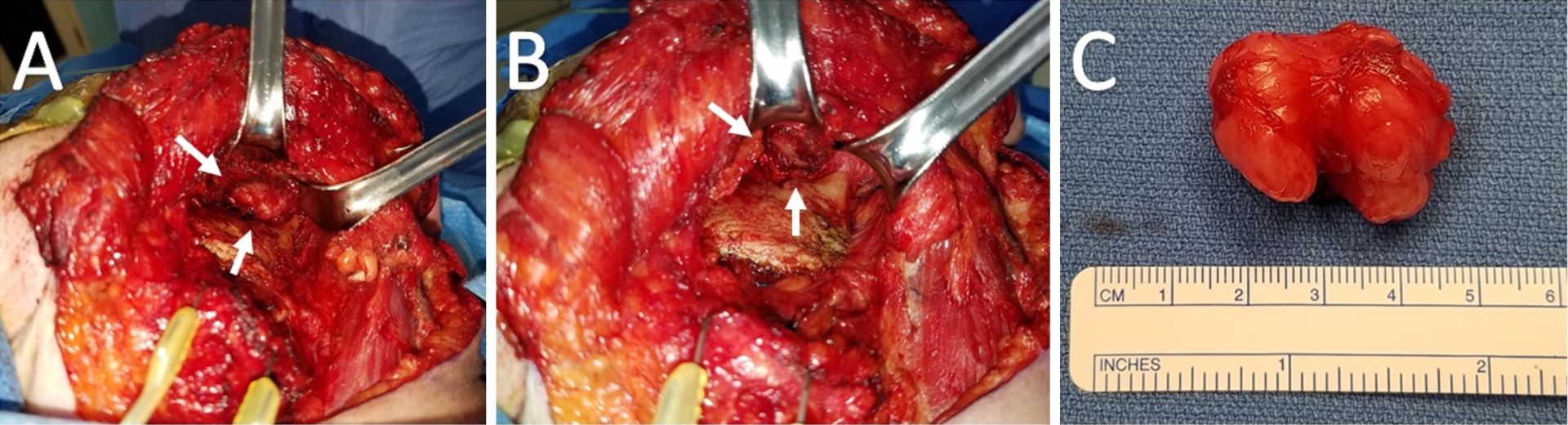
Figure 2. (A) Intraoperative view of the left infratemporal fossa from a transcervical approach. The tumor is saddling the posterior coronoid process and superior to the sigmoid notch. (B) A residual well-defined pocket status post-resection of the tumor and coronoid process. (C) Gross specimen measuring 3.4 x 2.9 x 2.7 cm with multiple lobulations and a smooth, glistening fibrous capsule. The central notch of the tumor demarcates the coronoid process upon which it cradled.
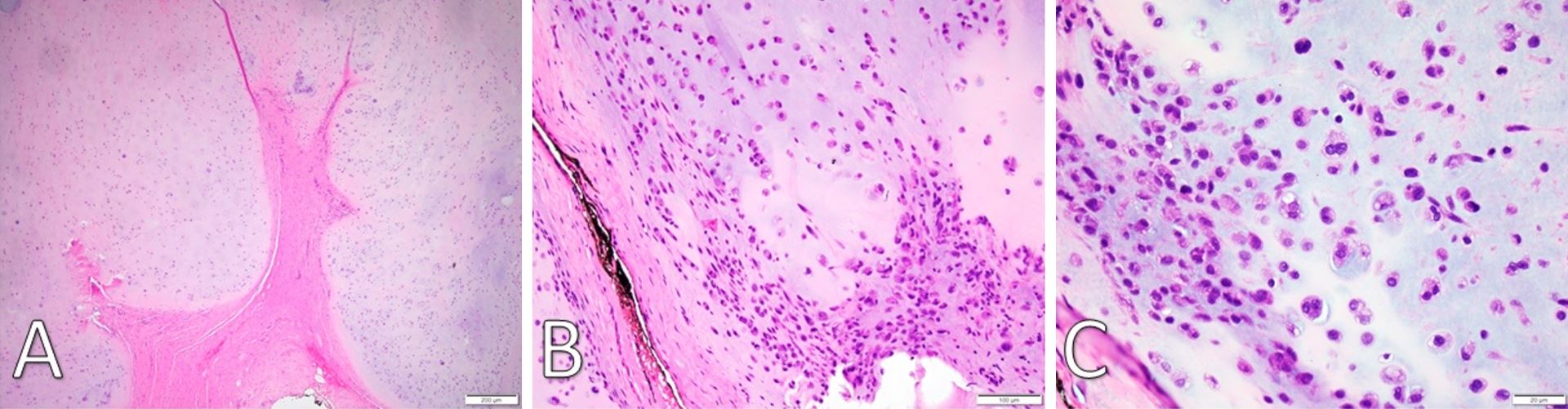
Figure 3. (A) H&E staining under low-power microscopy (4x) of a well-demarcated fibromyxoid capsule forming septa into hyaline cartilage and creating clear lobulations. (B) H&E staining (20x) displays areas of increased cellularity and nuclear irregularity within the hyaline cartilage. The fibromyxoid septation is poorly cellular, lacking vascular infiltration, and partially infiltrated by spindle-shaped chondroid cells. (C) H&E staining under high-power microscopy (40x) identifies a grey-blue chondroid matrix. Some tumor cells are found within lacunae. There is marked hyperchromatism and nuclear irregularity with some binucleate forms. Occasional spindle cells within the chondroid are seen. H&E, Hemotoxylin and Eosin.
Systematic Review
This review was performed in accordance to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) reporting standards [11]. The inclusion and exclusion criteria for eligibility were established prior to conducting the literature search. Studies were included for review if they (1) examined juxtacortical (parosteal, periosteal) chondrosarcomas; (2) involved the head and neck region; (3) included patient characteristics; (4) described medical history, duration and character of symptoms, and physical exam findings; (5) reported tumor characteristics, treatment modalities, and histological analysis; (6) utilized and included imaging findings; (7) described patient follow-up status and treatment outcomes.
Reports were excluded if they (1) did not examine a human subject; (2) did not investigate a juxtacortical (parosteal or periosteal) chondrosarcoma; (3) did not involve the head and neck region; (4) lacked sufficient patient, tumor, or imaging data; (5) did not publish in the English language; (6) were a review of literature. Eligible studies were identified utilizing a methodical and exhaustive search of all prior publications to February 15, 2019, located within the PubMed, MEDLINE, Embase, and Web of Science online databases. The MeSH terms “skull neoplasms”, “chondrosarcoma”, and “head and neck neoplasms” were cross-referenced with the terms “juxtacortical”, “parosteal”, and “periosteal” using Boolean logic. Additionally, the specific search terms “periosteal chondrosarcoma”, “juxtacortical chondrosarcoma”, and “parosteal chondrosarcoma” were utilized.
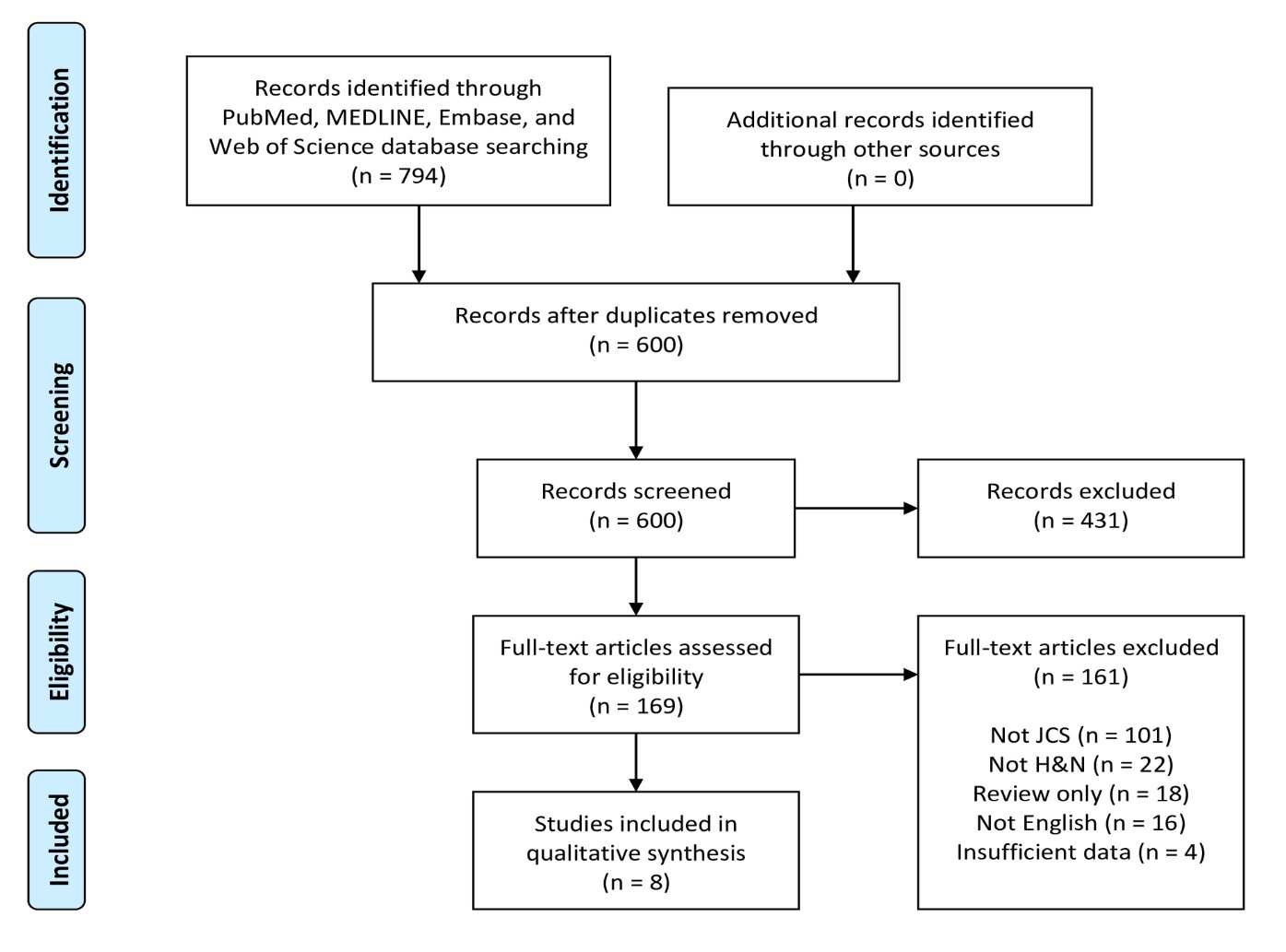
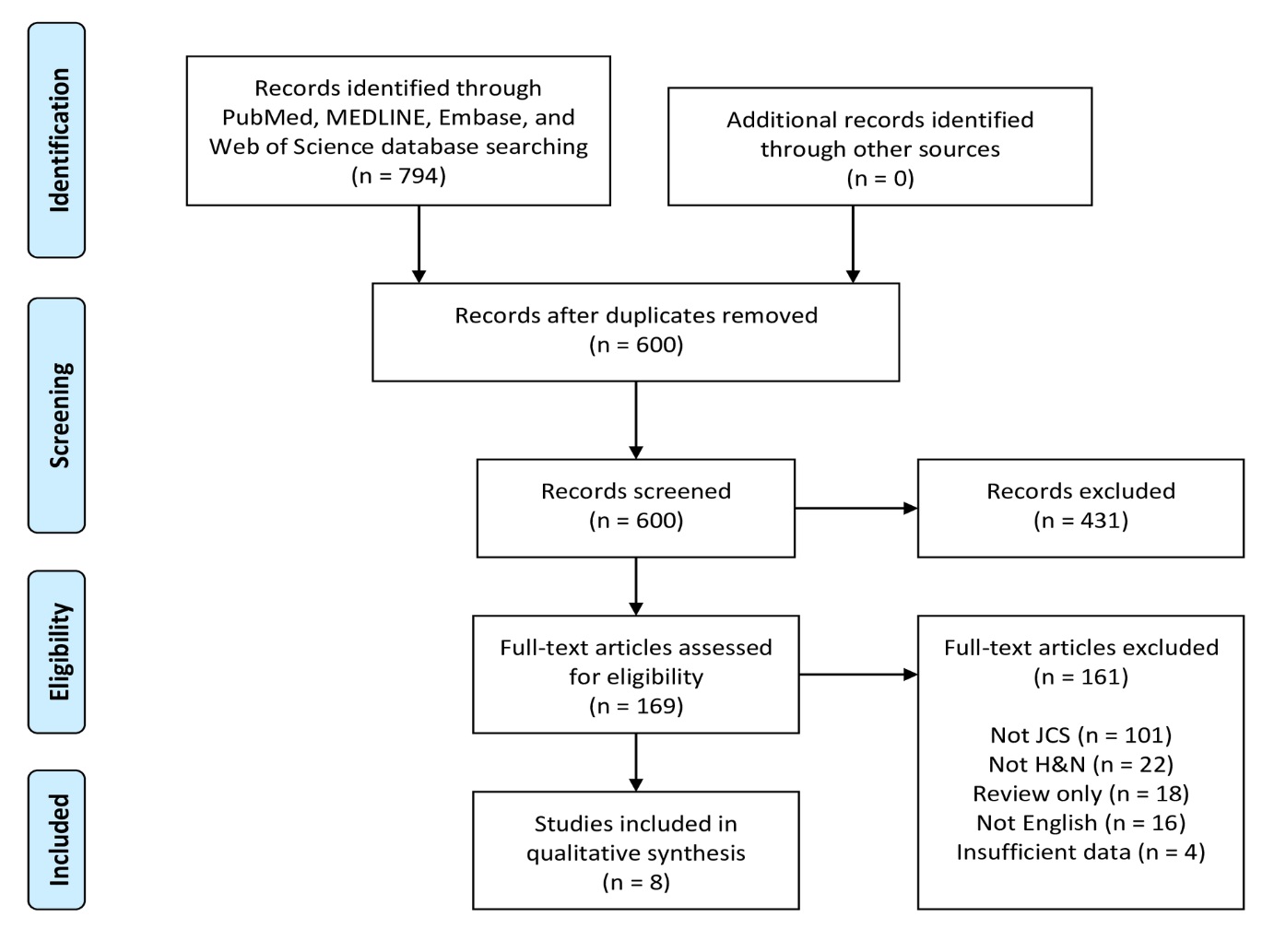
Figure 4. PRISMA flow diagram of article identification and selection. H&N, head and neck; JCS, juxtacortical chondrosarcoma; PRISMA, Preferred Reporting Items for Systematic Reviews and Meta-Analyses.
Study Selection
A flowchart of the systematic review selection process is presented in Figure 4. The PubMed, MELDINE, Embase, and Web of Science databases yielded 469, 56, 140 and 129 records, respectively, for a total of 794 articles. After removal of 194 duplicates, 600 abstracts remained. Of these initial records, 431 were excluded after title and abstract screening. The remaining 169 articles were subjected to full-text evaluation. After comprehensive review, 161 articles were excluded, leaving 8 articles as eligible for final inclusion in this systematic review [12-19].
Demographics and Clinical Findings
The publication dates ranged from 1995-2018 for a total of eight patients. The aforementioned case report constitutes the ninth documented case of HNJCS and was included as part of the data set. A summary of all clinical findings is found in Table 1. The cases consisted of six males (66.7%) and three females (33.3%) with ages ranging from 23-66 years with a mean and median of 41.2 and 41 years, respectively. Past medical history was not recorded for five cases, one patient was 31 weeks pregnant, one history was “noncontributory,” two patients had significant smoking histories, and one had prior chemotherapy for Hodgkin’s lymphoma. Reported symptoms included painless swelling (n = 7), tooth mobility (n = 2), impaired painful jaw mobility (n = 1), and mid-facial pain (n = 1). Duration of symptoms prior to presentation ranged from 1 to 84 months with a median of 2.5 months. For lesions that were palpable on clinical examination (n = 6), a firm, non-tender, fixed swelling to the underlying bone was noted; three cases reported ulcerations of the lesion. Seven cases identified the tumor emanating from the mandible, one from the hyoid bone, and one from the anterior maxilla. Of the mandibular lesions, three were identified at the symphysis and one each at the condyle, coronoid process, angle, and body.
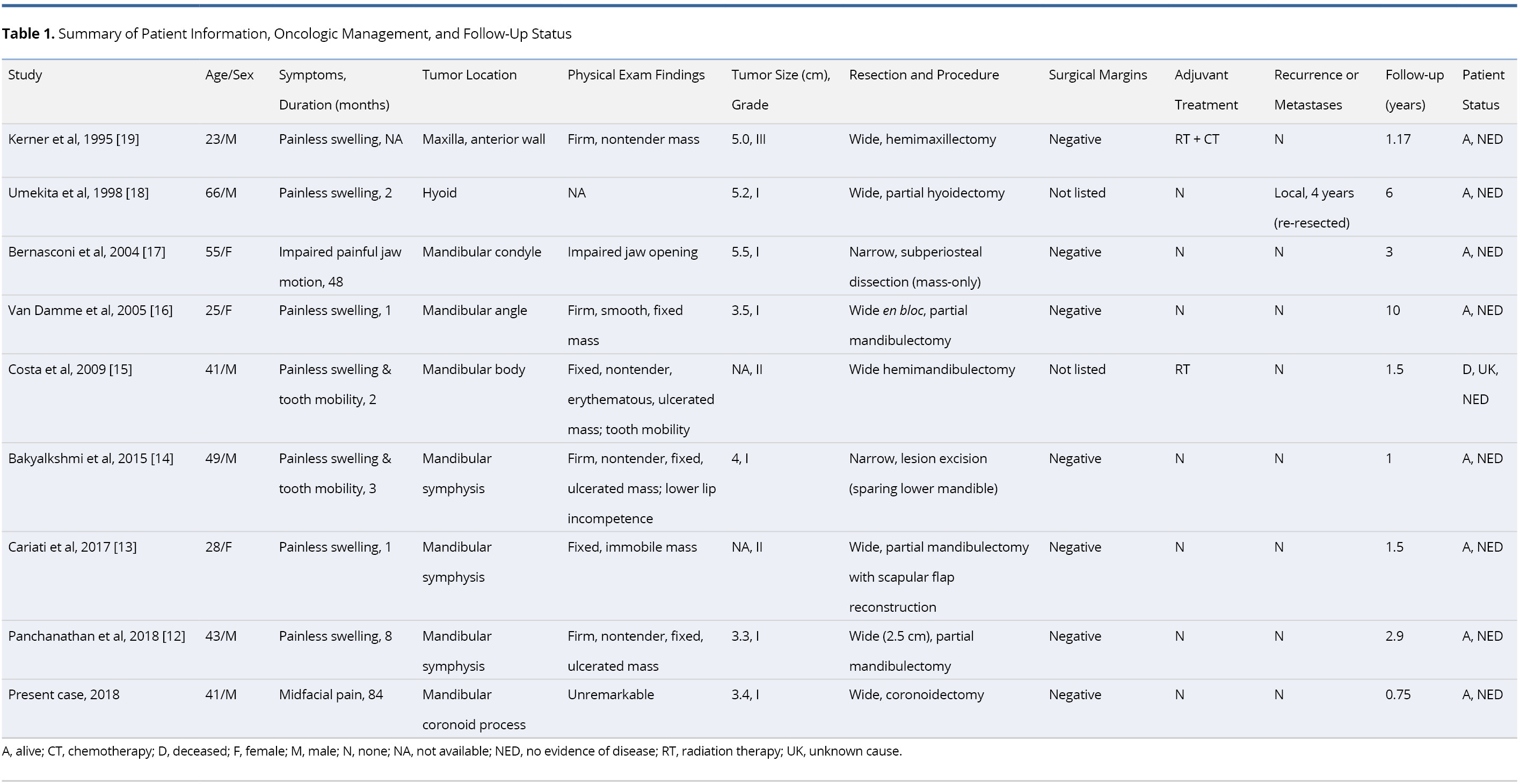
Imaging and Histopathologic Findings
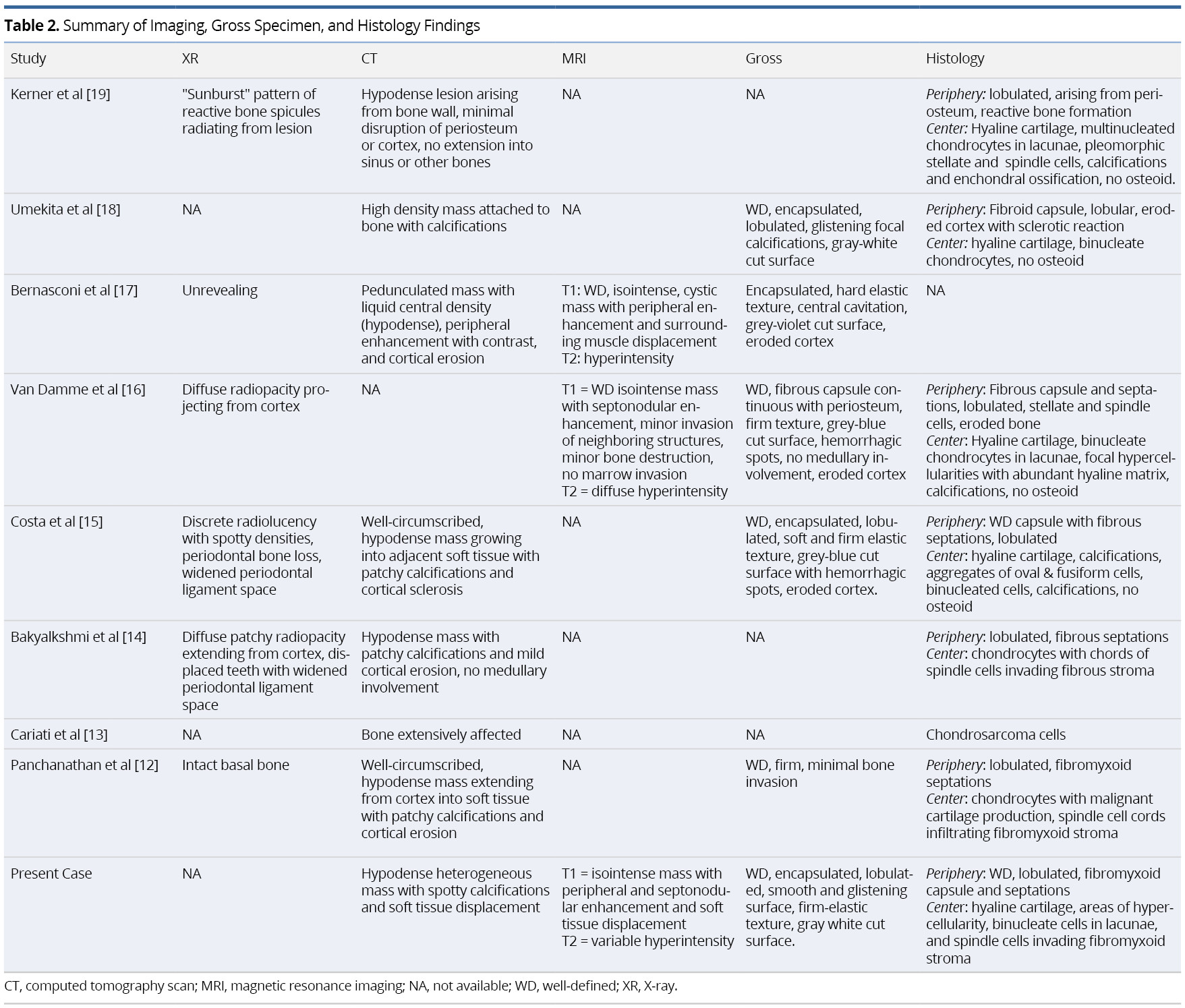
Preoperative imaging included X-ray (XR) orthopantomographs (n = 5), computed tomography (n = 8), magnetic resonance imaging (n = 3), and Technetium-99m (99mTc) scintigraphy (n = 2). One XR was taken of the removed specimen en bloc [19]. Initial histopathologic sampling was performed using incisional biopsy (n = 6) and/or fine needle biopsy (n = 3). Individual imaging and pathologic findings are summarized in Table 2.
Tumor Characteristics and Management Outcomes
Tumor information and management outcomes are summarized in Table 1. TNM staging was assigned to cases in accordance to the eighth edition of the American Joint Committee on Cancer (AJCC) Staging Manual of bone tumors [20]. The average maximum diameter of the resected specimen was 4.3 cm, ranging from 3.3-5.2 cm. There were no occurrences of nodal or metastatic disease. Six cases reported low-grade (Grade I), two reported intermediate-grade (Grade II) and one reported high-grade (Grade III). All cases were T1 or Tx with no nodal (N0) or metastatic (M0 or Mx) disease identified.
All patients initially underwent surgical treatment with wide-margin resection (n = 7) or narrow-margin resection (n = 2). Margins were negative of tumor in seven cases and not specified in two. One case underwent adjuvant radiotherapy, and another underwent both adjuvant radiation and chemotherapy. One patient had local recurrence four years after initial resection, which was managed with additional resection. There were no reported distal metastases after initial treatment. None of the patients had evidence of disease at their last documented follow-up visit, although one patient had deceased from unknown cause. Median follow-up was 1.5 years, ranging from 0.75-10 years.
Chondrosarcoma was first recognized as a distinct histopathologic entity in 1939 [21]. Prior to this, chondrosarcoma was classified as osteogenic sarcoma. Lichtenstein was the first to identify and define “periosteal” malignancies in 1955, noting to have observed one periosteal chondrosarcoma in the upper humerus [22]. In 1958, Jaffe officially described the first “juxtacortical chondrosarcoma” in an elderly female [23]. Since then, all recorded JCS were documented outside of the H&N until 1995 [19].
As stated previously, the incidence of HNJCS remains exceedingly rare. Our systematic review of literature revealed only eight adequately reported cases of HNJCS, with our case constituting the ninth report. Excluded from our review were two cases of unspecified and undescribed HNJCS by Koch et al after their examination of 400 head and neck conventional chondrosarcomas (HNCCS) cases in the American College of Surgeons’ National Cancer Data Base (NCDB) comprising 355,019 H&N cancer cases [4]. The largest study to-date of JCS was by Goedhart et al in 2014, which examined 17,522 cases over 59 years of all bone tumors in the Netherlands. The study identified 1,791 total cases of CS out of which only 36 were JCS (2.0%), none of which were in the head and neck [8]. Papagelopoulos et al examined 11,807 reports spanning 80 years of pathology records at the Mayo Clinic and found 24 JCS records, none of which affected the head and neck [10]. Ellis et al examined the Surveillance, Epidemiology, and End Results (SEER) database in 2016, specifically comparing HNCCS to CS everywhere else [3]. Only 44 cases of JCS were identified, none of which were in the head and neck.
Our study identified a median age of 41 years for HNJCS with a 66.7% male preponderance. Larger JCS studies have reported average ages ranging from 33.6-37.6 years [8,10]. Although HNCCS also exhibit male preponderance, HNCCS have been found to affect an older population with a median age of 51.0 years [4]. Estrogen has been shown to inhibit the differentiation of the periosteal progenitor cells while androgens stimulate this differentiation [24,25]. This growth and differentiation response of the perichondrium to androgenic hormones may explain the slight male predominance seen in both JCS as well as CCS [8,10].
The signs and symptoms of HNJCS seem rather indolent, often presenting as a painless swelling over several months to years. Pain associated with HNJCS was only observed in two patients in whom the mass occupied the infratemporal fossa. We cannot adequately justify whether the pain is from malignant inflammation or that of nerve compression at the infratemporal fossa.
Although the use of XR imaging is useful in juxtacortical chondrosarcomas elsewhere (JCE), particularly the appendicular skeleton [8], this modality of imaging provided nonspecific findings with little diagnostic relevance in our review of HNJCS cases. CT findings demonstrated hypodense lesions originating from the bone surface with spotty calcifications and cortical reactions. MRI is instrumental in identifying soft tissue involvement as well as medullary extension of the tumor, which has prognostic and surgical implications [26]. HNJCS appear as lobulated iso- or hypointense on T1W images with non-uniform peripheral and septal enhancement after contrast. T2W images exhibit heterogeneous hyperintense lesions. Fat-suppressed MRIs with contrast may augment imaging for sarcomas in obscured locations (such as the infratemporal fossa) [20]. Positron Emission Tomography (PET) with fluorodeoxyglucose (FDG) has been found useful in classifying cartilaginous neoplasms as benign or malignant, with maximum standard uptake values (SUVmax) generally <2.0 for benign tumors and ≥4.4 for intermediate- or high-grade tumors [27,28]. Given that none of the cases in our review received PET scans for diagnostic imaging, the role of this imaging modality remains uncertain in patients with HNJCS.
Most similar in content and appearance to low-grade JCS is the juxtacortical chondroma [26]. Although rare, it is more common than JCS and has nearly identical imaging and histological presentation to JCS, but generally appears in younger patients as a painless mass with diameter <3 cm [29]. It lacks endosteal scalloping on CT or cortical enhancement on MRI and shows radiotracer uptake <2.0 SUV on PET/CT [10,26]. Discriminating these from JCS histologically is difficult, but these tumors lack the atypia and hyper-cellularity of JCS and have a well-demarcated surrounding of lamellar bone [10]. Correct distinction between JCS and juxtacortical chondroma is critical as the latter does not have malignant or metastatic potential, and therefore may be managed conservatively [26].
Similarly, histopathologic differentiation of JCS from CCS requires astute microscopic examination due to several common features [15]. In both instances, lobules of hyaline cartilage with irregular distribution of cells are present with tumor cells often exhibiting nuclear pleomorphisms. Calcification within the hyaline matrix and cortical invasion are also common findings, but no malignant osteoid or bone are present within the tumor [10,15]. Both CCS and JCS may reveal a fibrous peripheral capsule with projecting septa that delve into the tumor, partitioning the cartilage into variable-sized lobules. The lobule margins contain undifferentiated mesenchymal spindle cells that invade the fibrous periphery [1]. The primary distinguishing feature of JCS, however, is the peripheral capsule is continuous with the surrounding periosteum [8,10]. Combining the clinical, radiological, and histological findings provide the greatest likelihood of accurate diagnosis. Other differentials to consider include: parosteal or periosteal osteosarcoma; periosteal Ewing’s sarcoma; juxtacortical dedifferentiated chondrosarcoma; and bizarre parosteal osteochondromatous proliferation (Nora’s lesion) [10].
JCS arise only at osseous sites, since they originate from the periosteum [8-10]. Seven of the nine documented case reports involved the mandible, with the remaining two found at the hyoid and maxilla. This contrasts HNCCS which are found affecting the laryngotracheal cartilages in addition to the bones of the skull and face [3,4].
The majority of cartilaginous neoplasms arise from bones that develop via endochondral ossification, which is the secondary ossification of a primary cartilage skeleton [24,30,31]. This is distinct from intramembranous ossification, seen in the craniofacial bones, where osseous tissue forms directly from neural crest-derived mesenchymal progenitors without an intermediate cartilage structure [31]. In turn, the development of the periosteum matches that of its underlying bone, being either endochondral or intramembranous [24,25]. Seven of the nine HNJCS were found at the distal and proximal aspects of the mandible or maxilla, all which undergo an endochondral-like ossification [32]. One case was identified at the mandibular body, which contrastingly undergoes intramembranous ossification [30]. The hyoid also undergoes an endochondral-like process via mesenchymal condensation and fusion with the 2nd and 3rd pharyngeal arches [33].
The above mechanism may explain the predilection for HNJCS at endochondrally-ossified locations. However, HNJCS have not been identified at the other endochondrally-derived skull bones (occipital, ethmoid, petrous temporal, and sphenoid) [34]. This perhaps is due to the absent mechanical-load bearing at the skull base compared to the maxilla, mandible and hyoid that endure mastication and swallowing forces. The periosteum development varies based upon several factors, including location, hormonal influence, age, and mechanical loading [24]. Load-bearing bones, as noted by JCS preponderance in the appendicular skeleton, have a substantially more active and developed periosteum than non-load-bearing. Furthermore, the multipotent capacity of periosteal mesenchymal cells into chondrocytes is readily seen in larger, load-bearing bones but absent in smaller, non-load bearing bones of the skull [24,25]. This may explain why HNJCS are yet to be observed within the skull base. It furthermore supports the previously-suggested hypothesis that JCS arise from aberrant differentiation and division of periosteal progenitors into the chondrogenic lineage [6]. However, an alternative hypothesis involves metaplasia of the periosteal fibroblasts into chondrocytes [35].
Bone sarcomas are graded as I (low), II (intermediate), or III (high) [20]. Six of nine (66.7%) HNJCS were low, two (22.2%) were intermediate, and one (11.1%) was identified as high. Papagelopoulos identified 75% of their JCE as low grade and 25% as intermediate [10]. Similarly, Goedhart identified 50% with low grade and 44% with intermediate grade, but none with high grade (6% unable to be evaluated) [8]. High grade was identified in one patient by Cleven et al. [9]. HNCCS have also shown to generally be low grade (50.2%) or intermediate grade (37.5%). However, 12.4% were found to be high grade or dedifferentiated [4]. Although grading is the most important aspect of CCS, it has not shown to be as predictive for future recurrence or metastases in JCS [36].
Primary surgical resection with wide margins (previously described as ‘extraperiosteal with normal bone margins’ [10]) was found to be the treatment modality of choice. In the setting of scarce supporting data, the therapeutic roles of radiation and chemotherapy remain unclear. Additionally, the role of proton beam therapy also remains unstudied, although certain reports have shown this to be an effective management modality in skull base CS [37]. As with several other neoplasms, a large majority of JCE recurrences are secondary to incomplete, marginal, or intralesional resections [8,10].
JCE appear to have excellent prognosis with extremely low rates of recurrence and metastases. A 5-year survival of >90% has been reported for patients who received wide, adequate surgical margins [8,10]. In our series, no metastases were found and only one case had local recurrence after four years. Following repeat resection, this patient was without disease at six years follow-up. Only one known death occurred, the etiology of which remains unknown. Although evidence-based data regarding optimal follow up of patient with HNJCS is lacking, reports of JCE have noted recurrence and distal metastases beyond five years [8-10].
To the best of our knowledge, this is the first systematic review of HNJCS documented in literature. We performed an expansive, thorough review of four large databases to identify all relevant articles. Our review is also the first to compare HNJCS to JCE and HNCCS. However, there are several limitations to our study. In addition to a small sample size, this report primarily analyzes observational case reports, which in turn are largely regarded as low-quality evidence [38]. The limited follow-up available for these malignancies limits evaluation of long-term overall and long-term disease-free survival. Given its rarity, the authors are also unable to recommend prospective trial designs to elucidate optimal management and follow up regimens.
HNJCS is an exceptionally rare clinical entity. HNJCS are often lower-grade, T1 malignancies with low rates of recurrence and metastases. Primary surgical excision with negative margins remains the therapeutic modality of choice. Therapeutic roles of radiotherapy and/or chemotherapy remain largely unstudied. Further, prospective studies are needed to provide evidence-based surveillance schedules for this cohort of patients.
Received date: February 04, 2019
Accepted date: March 07, 2019
Published date: March 14, 2019
This manuscript was presented as a poster at the Triological Society 2019 Combined Sections Meeting in Coronado, CA, January 24-26, 2019.
The study is in accordance with the ethical standards of the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
The study did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The authors report no financial or other conflict of interest relevant to this article, which is the intellectual property of the authors.
© 2019 The Author (s). This is an open-access article distributed under the terms of the Creative Commons Attribution 4.0 International License (CC-BY).
The study aims to assess the predictive values of certain psychological factors on the quality of life in patients with Head and Neck Cancer after radiotherapy. The authors conclude that the identification and the understanding of the depressive symptoms of patients, their beliefs about their illness as well as their coping strategies may provide the basis for timely implementation of appropriate intervention that may improve the quality of life in patients.
The clinical significance of otitis media with effusion (OME), a complication associated with cleft lip/palate (CLP), is often overlooked in children. The author reviews the pathogenesis, clinical manifestations, and diagnoses of OME in children with CLP as well as the controversies surrounding treatment. He also provides a flowchart to guide the management of OME in children with CLP.
Traditionally, suturing techniques have been the mainstay for microvascular anastomoses, but owing to its technical difficulty and labour intensity, considerable work has gone into the development of sutureless microvascular anastomoses. In this review, the authors take a brief look at the developments of this technology through the years, with a focus on the more recent developments of laser-assisted vascular anastomoses, the unilink system, vascular closure staples, tissue adhesives, and magnets. Their working principles, with what has been found concerning their advantages and disadvantages are discussed.
Epistaxis (i.e., nosebleed) is a common otolaryngologic emergency; however, it is seldom life-threatening and most minor nosebleeds stop on their own or under primary care from medical staff. Nonetheless, cases of recurrent epistaxis should be checked by an otolaryngologist, and severe nosebleeds should be referred to the emergency department to avoid adverse consequences, including hypovolemic shock or death. This paper reviews current advances in our understanding of epistaxis as well as updated treatment algorithms to assist clinicians in optimizing outcomes.
This article presents a comprehensive review of schwannomatosis affecting cranial nerves, delineating its unique characteristics distinct from other forms of neurofibromatosis. By addressing diagnostic complexities and the evolving criteria for identification, the paper emphasizes the critical need for accurate recognition of schwannomatosis to facilitate effective management and provide essential genetic counseling. Enriched with a detailed case study, this review delivers vital insights into the epidemiology, symptomatology, and therapeutic strategies for schwannomatosis, advocating for a revision in current clinical approaches. This work is indispensable for medical professionals aiming to enhance diagnostic precision, comprehend genetic underpinnings, and improve patient outcomes. Offering a thorough analysis of this rare condition, the article is pivotal not only for clinicians and researchers in the neurogenetic field but also for a broader spectrum of medical and scientific communities, bridging a notable gap in contemporary medical literature.
The author reframes review articles as evidence infrastructure that updates clinical consensus and identifies research gaps. The author argues that narrative reviews integrate findings across biological scales and translate molecular data into clinically meaningful explanations of disease behavior. Using cholesteatoma, the author illustrates how molecular evidence can recast invasiveness and recurrence into a mechanistic model that also motivates the pursuit of nonsurgical therapies. The author contrasts this interpretive role with systematic reviews, which function as reproducible decision chains grounded in transparent, verifiable methodology. Finally, the author cautions that AI assisted automation can propagate error unless it is governed by expert oversight within a human in the loop model that preserves auditable decision chains.
Jones AJ, Alwani M, Summerlin DJ, Sim MW. Head and neck juxtacortical chondrosarcoma: A systematic review. Arch Otorhinolaryngol Head Neck Surg 2019;3(1):4. https://doi.org/10.24983/scitemed.aohns.2019.00107
 PDF
PDF